400-998-5282
专注多肽 服务科研
400-998-5282
专注多肽 服务科研
研究表明,Amyloid β-Protein (17-21)在淀粉样变的大鼠模型中,β-Sheet Breaker Peptide显著减少体内β淀粉样蛋白的沉积,完全阻止淀粉样蛋白原纤维的形成。
编号:110189
CAS号:182912-74-9/2763585-09-5
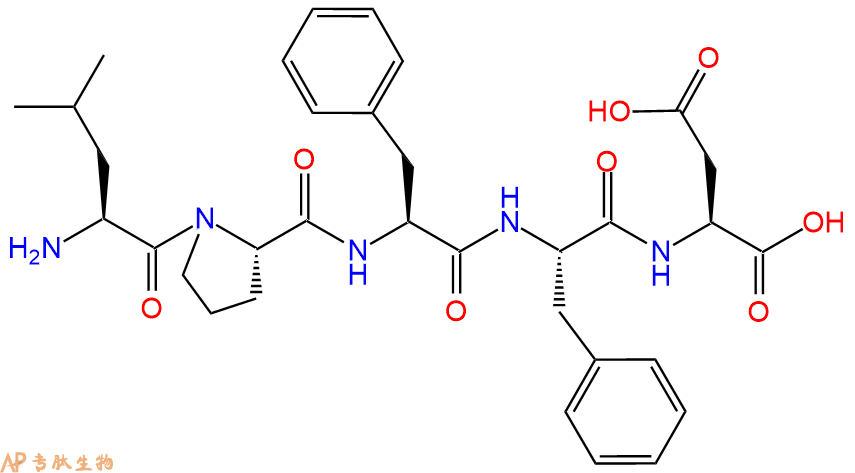
单字母:H2N-LPFFD-OH
The pentapeptide LPFFD (iAβ5), a highly efficient β-sheet breaker, inhibited amyloid formation in vitro, prevented amyloid neurotoxicity in cell culture, reduced in vivo cerebral Aβ deposition and completely blocked amyloid fibril formation in rat brain.
β-Sheet Breaker Peptide iAβ5的化学式为C33H43N5O8,多肽序列为H-Leu-Pro-Phe-Phe-Asp-OH。在大鼠模型中,它能抑制淀粉样蛋白的生成。研究表明,在淀粉样变的大鼠模型中,β-Sheet Breaker Peptide显著减少体内β淀粉样蛋白的沉积,完全阻止淀粉样蛋白原纤维的形成。目前的证据表明,β-淀粉样蛋白沉积或Aβ中间体在阿尔茨海默氏病(AD)患者的发病机制中发挥重要作用。β-Sheet Breaker Peptide可以抑制脑中β淀粉样蛋白的沉积,是治疗阿尔茨海默氏病的重要靶标。
β5抑制大鼠脑模型中淀粉样蛋白的形成。
Beta-Sheet Breaker Peptide iAβ5 inhibits amyloid formation in rat brain models.
β-折叠破坏肽iAß5(散装)是一种抑制蛋白质中β-折叠形成的肽。它已被证明是蛋白质相互作用的优秀抑制剂,对其靶标具有良好的选择性。该肽也具有高纯度,可用作研究离子通道和抗体功能的研究工具。\n\n6-氟-3-吲哚氧基-β-D-吡喃半乳糖苷:利福喷丁是一种属于利福霉素类的抗结核药物。它是治疗结核病最活跃的利福霉素。利福喷丁通过与DNA依赖性RNA聚合酶结合来抑制细菌生长,从而阻止转录和复制。使用膜片钳技术对人红细胞显示出人类活动的高频率。
β-Sheet Breaker Peptide iAß5 (Bulk) is a peptide that inhibits the formation of β-sheets in proteins. It has been shown to be an excellent inhibitor of protein interactions, with good selectivity for its target. This peptide also has high purity and can be used as a research tool for studying the function of ion channels and antibodies.\n\n6-Fluoro-3-indoxyl-beta-D-galactopyranoside: Rifapentine is an anti-tuberculosis drug that belongs to the class of rifamycins. It is the most active of the rifamycins for the treatment of tuberculosis. Rifapentine inhibits bacterial growth by binding to DNA-dependent RNA polymerase, thereby preventing transcription and replication. The high frequency of human activity has been shown using a patch-clamp technique on human erythrocytes.
β折叠破坏肽iAß5是一种与淀粉样β蛋白相互作用以破坏β折叠形成的肽。它已被证明可以抑制淀粉样β蛋白聚集成低聚物和原纤维,并破坏淀粉样β蛋白自组装成各种构象。β-折叠破坏肽iAß5是淀粉样β蛋白上受体和激活剂结合位点的配体,抑制这些位点的相互作用。这抑制了与阿尔茨海默病相关的下游信号通路的激活。
Beta-Sheet Breaker Peptide iAß5 is a peptide that interacts with amyloid beta proteins to disrupt the formation of beta sheets. It has been shown to inhibit the aggregation of amyloid beta proteins into oligomers and fibrils, and disrupts the self-assembly of amyloid beta protein into various conformations. Beta-Sheet Breaker Peptide iAß5 is a ligand for both receptor and activator binding sites on amyloid beta proteins, inhibiting the interaction of these sites. This inhibits the activation of downstream signalling pathways associated with Alzheimers disease.
在体外抑制淀粉样蛋白形成并防止细胞培养中淀粉样蛋白神经毒性的五肽。它还具有减少体内脑Abeta沉积并完全阻断大鼠脑中淀粉样蛋白原纤维形成的能力。
Pentapeptide that inhibited amyloid formation in vitro and prevented amyloid neurotoxicity in cell culture. It also has the capacity to reduce in vivo cerebral Abeta deposition and to completely block amyloid fibril formation in rat brain.
淀粉肽背景:β淀粉样蛋白(Aβ或Abeta)是从淀粉样前体蛋白加工而成的含有36–43个氨基酸的多肽。Aβ是与阿尔兹海默病相关的淀粉样蛋白斑的成分。已有证据表明,Aβ是一个多功能肽,具有显著的非病理性活性。Aβ是阿尔兹海默病患者脑中发现的沉积物的主要成分。在散发性阿尔兹海默病患者的脑中,Aβ的水平升高,造成脑血管病变和神经毒性。Aβ蛋白是由β和γ分泌酶的连续作用而产生的。γ分泌酶产生Aβ肽的C末端,在APP的转膜结构域切割,可以产生许多36-43个氨基酸残基长度的异构体,最常见的异构体是Aβ40和Aβ42。更长形式的Aβ在内质网中切割产生,而更短形式的Aβ在反面高尔基网中产生。
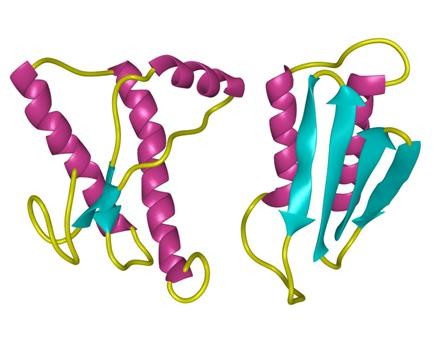
structure of Amyloid β-Peptide (1-40) (human)
淀粉样蛋白肽的 定义淀粉样蛋白 是丝状蛋白质沉积物,大小从纳米到微米不等,并且由肽β链的平行或反平行排列形成的聚集的肽β折叠构成。
结构特征:使用固态NMR(SSNMR),与计算能量最小化过程结合,Tycko和合作者已经提出从淀粉状蛋白肽SS(Aß1-40)的40个残基的形式形成的淀粉样蛋白原纤维的结构在pH 7.4和24 o C在静止条件下。在这种结构中,每个Aß1-40分子在原纤维的核心区域贡献一对ß链,大约跨越残基12-24和30-40。这些由回路25-29连接的链不是同一张ß-sheet的一部分,但参与同一原丝内两个不同的ß-sheets的形成。不同的Aß分子2、3至少从第9到39位残基以平行排列和对齐的方式相互堆叠。通过调用其他实验约束,例如使用透射电子显微镜(TEM)观察到的原丝直径和单位质量通过扫描透射电子显微镜(STEM)1、2测得的长度表明,单个原丝是由四个ß片组成的,它们之间的距离约为10Å。
作用模式:阿尔茨海默氏病(AD)是淀粉样蛋白丝状沉积物的结果,淀粉状蛋白沉积物在分子水平上定义该疾病,发生在神经周膜,轴突,树突和神经元末端,如神经原纤维缠结(NFT),在细胞外神经纤维中淀粉样斑块(APC),以及周围的血管称为淀粉样嗜血性血管病(ACA)。淀粉样蛋白沉积物显然发生在发展NFT的神经元末端区域。已经表明,APC和ACA的主要成分已被证明是4.5kDa的淀粉样蛋白,最初被称为“β-蛋白”或“淀粉样蛋白A4”,我们现在将其称为“βA4”。
功能:钙失调和膜破坏是可溶性淀粉样蛋白低聚物普遍存在的神经毒性机制:进行了一项研究,以研究Ca 2+信号转导可能参与淀粉样蛋白诱导的细胞毒性,疾病相关淀粉样蛋白(β,病毒,胰岛淀粉样蛋白)的均质制剂制备了处于各种聚集状态的多肽,聚谷氨酰胺和溶菌酶),并测试了它们对加载fluo-3的SH-SY5Y细胞的作用。寡聚形式的所有淀粉样蛋白的应用(0.6-6 µg / ml)迅速(约5 s)使细胞内Ca 2+升高,而等量的单体和原纤维则没有。细胞内Ca 2+耗尽后,Abeta42低聚物引起的Ca 2+信号持续存在店,和小信号仍留在钙2 + -游离介质,指示从细胞外和细胞内Ca贡献2+源。膜对Ca 2+的渗透性增加不能归因于内源性Ca 2+通道的活化,因为反应不受强力的Ca 2 +-通道阻滞剂钴的影响。取而代之的是,观察到Abeta42和其他低聚物引起阴离子荧光染料的快速细胞泄漏,这表明膜通透性普遍提高。导致的离子和分子通量失调可能为许多淀粉样变性疾病中Ca 2+失调提供了由低聚物介导的毒性的常见机制。离子起着至关重要的作用,因为它们的跨膜浓度梯度很强,并且参与了细胞功能障碍和死亡。
2型糖尿病中的胰岛淀粉样蛋白和毒性低聚物假说: 2型糖尿病(T2DM)的特征是胰岛素抵抗,胰岛素分泌缺陷,β细胞量减少,β细胞凋亡增加和胰岛淀粉样蛋白。胰岛淀粉样蛋白源自胰岛淀粉样蛋白多肽(IAPP,胰岛淀粉样多肽),该蛋白是通过胰β细胞与胰岛素共表达和共分泌的蛋白。与其他淀粉样蛋白一样,IAPP具有形成膜渗透性毒性低聚物的倾向。越来越多的证据表明,这些有毒的寡聚体而不是这些蛋白质的细胞外淀粉样蛋白形式,是导致神经退行性疾病中神经元丢失的原因。有人提出,胞内IAPP寡聚物的形成可能会导致T2DM 6中的β细胞丢失。
定义
酶是用于生化反应的非常有效的催化剂。它们通过提供较低活化能的替代反应途径来加快反应速度。酶作用于底物并产生产物。一些物质降低或什至停止酶的催化活性被称为抑制剂。
发现
1965年,Umezawa H分析了微生物产生的酶抑制剂,并分离出了抑制亮肽素和抗痛药的胰蛋白酶和木瓜蛋白酶,乳糜蛋白酶抑制的胰凝乳蛋白酶,胃蛋白酶抑制素抑制胃蛋白酶,泛磷酰胺抑制唾液酸酶,乌藤酮抑制酪氨酸羟化酶,多巴汀抑制多巴胺3-羟硫基嘧啶和多巴胺3-羟色胺酶酪氨酸羟化酶和多巴胺J3-羟化酶。最近,一种替代方法已应用于预测新的抑制剂:合理的药物设计使用酶活性位点的三维结构来预测哪些分子可能是抑制剂1。已经开发了用于识别酶抑制剂的基于计算机的方法,例如分子力学和分子对接。
结构特征
已经确定了许多抑制剂的晶体结构。已经确定了三种与凝血酶复合的高效且选择性的低分子量刚性肽醛醛抑制剂的晶体结构。这三种抑制剂全部在P3位置具有一个新的内酰胺部分,而对胰蛋白酶选择性最高的两种抑制剂在P1位置具有一个与S1特异性位点结合的胍基哌啶基。凝血酶的抑制动力学从慢到快变化,而对于胰蛋白酶,抑制的动力学在所有情况下都快。根据两步机理2中稳定过渡态络合物的缓慢形成来检验动力学。
埃米尔•菲舍尔(Emil Fischer)在1894年提出,酶和底物都具有特定的互补几何形状,彼此恰好契合。这称为“锁和钥匙”模型3。丹尼尔·科什兰(Daniel Koshland)提出了诱导拟合模型,其中底物和酶是相当灵活的结构,当底物与酶4相互作用时,活性位点通过与底物的相互作用不断重塑。
在众多生物活性肽的成熟过程中,需要由其谷氨酰胺(或谷氨酰胺)前体形成N末端焦谷氨酸(pGlu)。游离形式并与底物和三种咪唑衍生抑制剂结合的人QC的结构揭示了类似于两个锌外肽酶的α/β支架,但有多个插入和缺失,特别是在活性位点区域。几种活性位点突变酶的结构分析为针对QC相关疾病5的抑制剂的合理设计提供了结构基础。
作用方式
酶是催化化学反应的蛋白质。酶与底物相互作用并将其转化为产物。抑制剂的结合可以阻止底物进入酶的活性位点和/或阻止酶催化其反应。抑制剂的种类繁多,包括:非特异性,不可逆,可逆-竞争性和非竞争性。可逆抑制剂 以非共价相互作用(例如疏水相互作用,氢键和离子键)与酶结合。非特异性抑制方法包括最终使酶的蛋白质部分变性并因此不可逆的任何物理或化学变化。特定抑制剂 对单一酶发挥作用。大多数毒药通过特异性抑制酶发挥作用。竞争性抑制剂是任何与底物的化学结构和分子几何结构非常相似的化合物。抑制剂可以在活性位点与酶相互作用,但是没有反应发生。非竞争性抑制剂是与酶相互作用但通常不在活性位点相互作用的物质。非竞争性抑制剂的净作用是改变酶的形状,从而改变活性位点,从而使底物不再能与酶相互作用而产生反应。非竞争性抑制剂通常是可逆的。不可逆抑制剂与酶形成牢固的共价键。这些抑制剂可以在活性位点附近或附近起作用。
功能
工业应用中, 酶在商业上被广泛使用,例如在洗涤剂,食品和酿造工业中。蛋白酶用于“生物”洗衣粉中,以加速蛋白质在诸如血液和鸡蛋等污渍中的分解。商业上使用酶的问题包括:它们是水溶性的,这使得它们难以回收,并且一些产物可以抑制酶的活性(反馈抑制)。
药物分子,许多药物分子都是酶抑制剂,药用酶抑制剂通常以其特异性和效力为特征。高度的特异性和效力表明该药物具有较少的副作用和较低的毒性。酶抑制剂在自然界中发现,并且也作为药理学和生物化学的一部分进行设计和生产6。
天然毒物 通常是酶抑制剂,已进化为保护植物或动物免受天敌的侵害。这些天然毒素包括一些已知最剧毒的化合物。
神经气体( 例如二异丙基氟磷酸酯(DFP))通过与丝氨酸的羟基反应生成酯,从而抑制了乙酰胆碱酯酶的活性位点。
参考
1、Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
2、Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
3、Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
4、Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
5、Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
6、Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
Definition
Enzymes are very efficient catalysts for biochemical reactions. They speed up reactions by providing an alternative reaction pathway of lower activation energy. Enzyme acts on substrate and gives rise to a product. Some substances reduce or even stop the catalytic activities of enzymes are called inhibitors.
Discovery
In 1965, Umezawa H analysed enzyme inhibitors produced by microorganisms and isolated leupeptin and antipain inhibiting trypsin and papain, chymostatin inhibiting chymotrypsin, pepstatin inhibiting pepsin, panosialin inhibiting sialidases, oudenone inhibiting tyrosine hydroxylase, dopastin inhibiting dopamine 3-hydroxylase, aquayamycin and chrothiomycin inhibiting tyrosine hydroxylase and dopamine J3-hydroxylase . Recently, an alternative approach has been applied to predict new inhibitors: rational drug design uses the three-dimensional structure of an enzyme's active site to predict which molecules might be inhibitors 1. Computer-based methods for identifying inhibitor for an enzyme have been developed, such as molecular mechanics and molecular docking.
Structural Characteristics
The crystal structures of many inhibitors have been determined. The crystal structures of three highly potent and selective low-molecular weight rigid peptidyl aldehyde inhibitors complexed with thrombin have been determined. All the three inhibitors have a novel lactam moiety at the P3 position, while the two with greatest trypsin selectivity have a guanidinopiperidyl group at the P1 position that binds in the S1 specificity site. The kinetics of inhibition vary from slow to fast with thrombin and are fast in all cases with trypsin. The kinetics are examined in terms of the slow formation of a stable transition-state complex in a two-step mechanism 2.
Emil Fischer in 1894 suggested that both the enzyme and the substrate possess specific complementary geometric shapes that fit exactly into one another.This is known as "the lock and key" model 3. Daniel Koshland suggested induced fit model where substrate and enzymes are rather flexible structures, the active site is continually reshaped by interactions with the substrate as the substrate interacts with the enzyme 4.
N-terminal pyroglutamate (pGlu) formation from its glutaminyl (or glutamyl) precursor is required in the maturation of numerous bioactive peptides. The structure of human QC in free form and bound to a substrate and three imidazole-derived inhibitors reveals an alpha/beta scaffold akin to that of two-zinc exopeptidases but with several insertions and deletions, particularly in the active-site region. The structural analyses of several active-site-mutant enzymes provide a structural basis for the rational design of inhibitors against QC-associated disorders 5.
Mode of Action
Enzymes are proteins that catalyze chemical reactions. Enzymes interact with substrate and convert them into products. Inhibitor binding can stop a substrate from entering the enzyme's active site and/or hinder the enzyme from catalyzing its reaction. There are a variety of types of inhibitors including: nonspecific, irreversible, reversible - competitive and noncompetitive. Reversible inhibitors bind to enzymes with non-covalent interactions like hydrophobic interactions, hydrogen bonds, and ionic bonds. Non-specific methods of inhibition include any physical or chemical changes which ultimately denature the protein portion of the enzyme and are therefore irreversible. Specific Inhibitors exert their effects upon a single enzyme. Most poisons work by specific inhibition of enzymes. A competitive inhibitor is any compound which closely resembles the chemical structure and molecular geometry of the substrate. The inhibitor may interact with the enzyme at the active site, but no reaction takes place. A noncompetitive inhibitor is a substance that interacts with the enzyme, but usually not at the active site. The net effect of a non competitive inhibitor is to change the shape of the enzyme and thus the active site, so that the substrate can no longer interact with the enzyme to give a reaction. Non competitive inhibitors are usually reversible. Irreversible Inhibitors form strong covalent bonds with an enzyme. These inhibitors may act at, near, or remote from the active site .
Functions
Industrial application, enzymes are widely used commercially, for example in the detergent, food and brewing industries. Protease enzymes are used in 'biological' washing powders to speed up the breakdown of proteins in stains like blood and egg. Problems using enzymes commercially include: they are water soluble which makes them hard to recover and some products can inhibit the enzyme activity (feedback inhibition) .
Drug molecules, many drug molecules are enzyme inhibitors and a medicinal enzyme inhibitor is usually characterized by its specificity and its potency. A high specificity and potency suggests that a drug will have fewer side effects and less toxic. Enzyme inhibitors are found in nature and are also designed and produced as part of pharmacology and biochemistry 6.
Natural poisons are often enzyme inhibitors that have evolved to defend a plant or animal against predators. These natural toxins include some of the most poisonous compounds known.
Nerve gases such as diisopropylfluorophosphate (DFP) inhibit the active site of acetylcholine esterase by reacting with the hydroxyl group of serine to make an ester.
References
Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
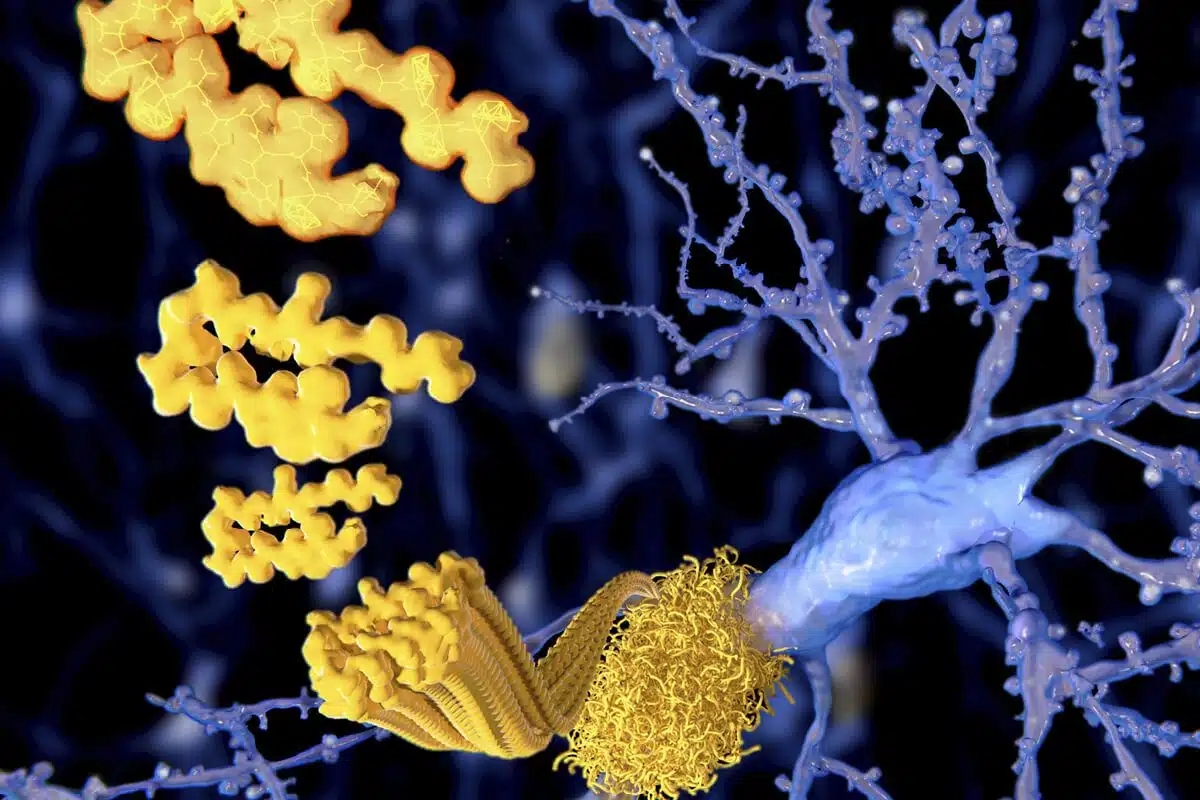
Extracellular amyloid-β peptide deposition into cerebellar plaques and formation of intracellular neurofibrillary fibers accompanied by the loss of neurons are characteristic histopathological lesions found in the brains of Alzheimer‘s disease patients. Individuals suffering from this disease show a gradual loss of cognitive functions and disturbances in behavior. Apart from some rare familial forms of the disease, the onset of Alzheimer‘s disease is usually above 60 years. Since the risk to develop the disease increases with age, Alzheimer‘s disease has turned into a major health and social problem in “first world” countries with an increasing proportion of older people, and is going to become one in emerging states. In this brochure we present amyloid peptides and related products for Alzheimer‘s disease research.
ALZHEIMER’S DISEASE
Alzheimer‘s disease (AD) is the prevalent cause of dementia in elderly people and has become one of the leading causes of death in developed countries together with cardiovascular disorders, cancer, and stroke. It is estimated that more than 46 millions of people suffer from AD all over the world. As age advances, the risk for developing AD increases. The frequency of AD at the age of 60-64 is about 1% and doubles approximately every five years. By the age of 90 and older, approximately 50% of the population suffers from this disease. AD is an irreversible and progressive neurodegenerative disorder. Symptoms include gradual loss of cognitive functions such as memory, verbal and visuospatial abilities, changes in personality, behavior, and activities of daily living. AD patients in the final stages are completely dependent on the care of others.
The characteristic lesions in the brains of AD patients were first described by the German neuropsychiatrist Alois Alzheimer in 1906 during the post-mortem examination of a mentally ill patient whose deterioration he had observed until her death. The lesions consisted of dense extracellular deposits, now designated as neuritic or senile plaques, and intracellular dense bundles of fibrils, which are now known as neurofibrillary tangles.
Currently, diagnosis of AD with adequate testing is approximately 90% accurate. It is based on the exclusion of a variety of diseases causing similar symptoms and a careful neurological and psychiatric examination, as well as neuropsychological testing. Imaging technologies for detecting amyloid plaques and tangles in vivo are becoming more precise and thus a valuable additional tool. Numerous potential biomarkers as α1 -antitrypsin, complement factor H, α2 -macroglobulin, apolipoprotein J, and apolipoprotein A-I for diagnosing AD are being evaluated. However, post-mortem histopathological examination of the brain is still the only definite diagnosis of this disease.
AD can be either inherited or sporadic. The inherited or familial AD is rare and comprises only 5-10% of all cases. Autosomal dominant mutations in the amyloid β/A4 protein precursor (APP) gene on chromosome 21 and the presenilin-1 or -2 genes on chromosomes 14 and 1, respectively, have been attributed to the early onset (before the age of 65) of this disease.
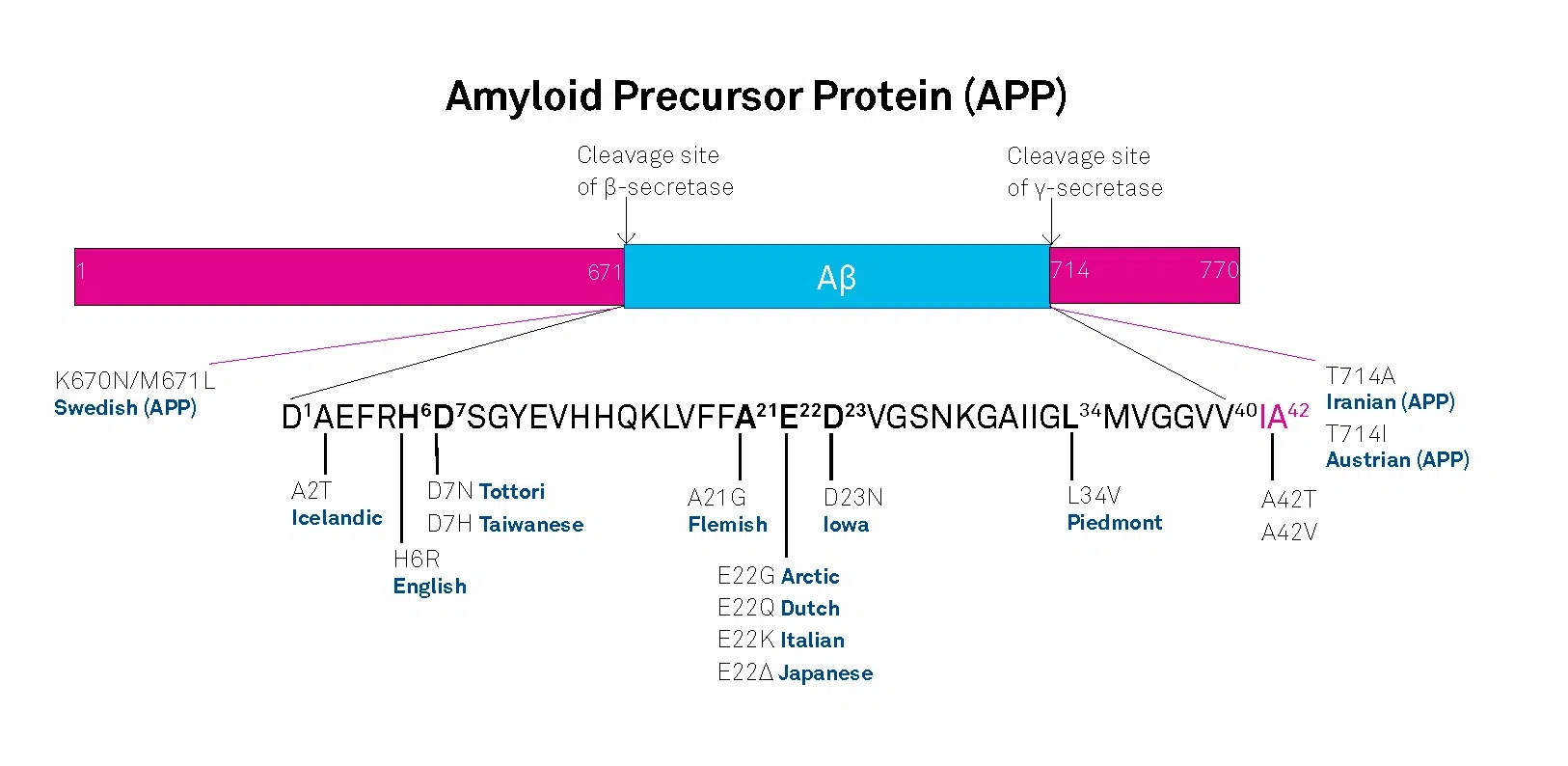
APP belongs to the type-1 integral membrane glycoproteins with at least 10 isoforms generated by alternative splicing of the 19 exons. The predominant transcripts are APP695, APP751, and APP770. A number of mutations within the APP gene have been detected in families with an inherited risk for early onset of AD. Usually, they are named after the region, in which they have been detected, e.g. the London APP717 mutations (V717I, V717F, V717G), the Swedish APP670/671 double mutation (K670N/M671L), the Flemish APP692 mutation (A692G), or the Dutch APP693 mutation (E693Q). The Swedish mutation of the β-secretase cleavage site of APP and mutations of positions 692-694 (Aβ 21-23), which strongly influence the aggregation behavior of Aβ, have been studied intensively.
A choice of relevant mutations in the Aβ region of APP is assembled in the table below.
| Exchanged Position in APP | Exchanged Position in Aβ | Designation |
|---|---|---|
| A673T | A2T | Icelandic |
| H677R | H6R | English |
| D678H | D7H | Taiwanese |
| D678N | D7N | Tottori |
| A692G | A21G | Flemish |
| E693D | E22∆ | Osaka |
| E693G | E22G | Arctic |
| E693Q | E22Q | Dutch |
| E693K | E22K | Italian |
| D694N | D23N | Iowa |
| L705V | L34V | Piedmont |
The presenilins are another group of proteins involved in the development of AD. Presenilins are integral membrane proteins with eight transmembrane domains localized in the endoplasmic reticulum and the Golgi apparatus. A multitude of mutations within the presenilin-1 and two within the presenilin-2 gene account for most of the cases of early onset of AD.
Genetic factors may contribute as well to the late onset of AD. Increased susceptibility is associated with the expression of different apolipoprotein E (ApoE) isoforms due to the polymorphism in the APOE gene on chromosome 19. In the central nervous system, ApoE has been implicated in growth and repair during development or after injury. Carriers of the APOEε4 allele show a higher risk in developing the disease than carriers of the other two possible alleles APOEε2 and APOEε3. The ApoEε4 effect seems to be dose-dependent since individuals with two of these alleles seem to be at two-fold higher risk to develop the disease than those with one allele. Polymorphisms of the α2 -macroglobulin gene on chromosome 12 and the gene coding low-density lipoprotein receptor-related protein 1 (LRP1), LRP1-C/T, have also been suggested to be a risk factor for the late onset of AD. However, further studies in this field are required.
A number of additional, most diverse risk factors have been proposed. These include gender, ethnic group, head trauma, cardiovascular diseases, and educational level.
AD THERAPEUTIC STRATEGIES RELY ON DETAILED KNOWLEDGE OF THE MOLECULES INVOLVED
Women, Hispanics, individuals who have experienced a head trauma earlier in life, and persons who suffer from cardiovascular diseases appear to have a higher risk of developing the disease.
The etiology of AD is still not completely understood. Initial research focused upon determining the molecular structure of the senile plaques and the neurofibrillary tangles originally described by Alois Alzheimer. The main constituents of the senile plaques were identified as cleavage products of APP, designated as amyloid β-peptides (Aβ peptides).
Depending on the composition and the fraction of fibrillar to non-fibrillar forms of these amyloid peptides, several kinds of senile plaques can be distinguished. Three types of proteases, α-secretase, β-secretase (or β-site APP-cleaving enzyme, BACE), and γ-secretase are involved in APP processing. APP can either be processed by the α- and γ- or by the β- and γ-secretases. The major two amyloid peptides identified in senile plaques, amyloid β-protein (1-40) (Aβ40) and amyloid β-protein (1-42) (Aβ42), are generated by successive proteolysis of APP by β- and γ-secretases. Cleavage of APP by β-secretase results in the release of the extracellular N-terminal protein fragment known as soluble APP-β molecule (sAPP-β). Then, the membrane-retained APP is further processed within the transmembrane domain by γ-secretase to yield either Aβ40 or Aβ42. The formation of Aβ40 and Aβ42 is a normal process, and both peptides can be detected in the plasma and cerebrospinal fluid (CSF) of healthy subjects.
In most studies, similar concentrations of Aβ40 have been measured in the CSF of both healthy controls and AD patients. On the other hand, Aβ42 concentrations in the CSF of AD patients are significantly lower than in normal controls, probably reflecting an increased deposition as insoluble plaques.
The neurofibrillary tangles found inside neurons of Alzheimer’s brains are composed of paired helical filaments whose main components are hyperphosphorylated forms of tau, a microtubule associated protein involved in promoting microtubule assembly and stabilization. Self-assembly into paired helical filaments is believed to be a result of hyperphosphorylation due to either the increased activity of protein kinases or the decreased activity of phosphatases.
Several lines of evidence support the view that the accumulation of Aβ42 in the brain is a primary event in the development of AD. Increased cerebral Aβ production appears to be characteristic for all the mutations within the APP and the presenilin genes of familial AD. In patients with Down syndrome (trisomy 21), elevated levels of APP and Aβ due to a third copy of the APP gene result in deposition of Aβ at an early age between 20 and 30.
Formation of neurofibrillary tangles is considered as a consequence of Aβ deposition with a further impact on the progression of the disease possibly due to disruption of axonal transport mechanisms in neurons.
The detailed knowledge about the molecules involved in AD has led to the development of several therapeutic strategies.
One strategy aims at the reduction of Aβ40 and Aβ42 by inhibition of either β- or γ-secretase activity or by clearance of Aβ in the brain by means of immunization with these peptides. Transition metals as Cu, Fe and Zn play an important role in the pathology of AD. Aggregation and neurotoxicity of Aβ are dependent on the presence of copper, so Cu-chelating agents showed promising effects in animal models. Another approach is the prevention of the cellular inflammatory response in the cerebral cortex elicited by the progressive accumulation of Aβ. Further preventive therapeutic strategies are based on the findings that cholesterol-lowering drugs such as statins and estrogen replacement therapy reduce the risk of developing AD. An additional treatment alternative would be the inhibition of the serine-threonine protein kinases, glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5), which are probably responsible for the phosphorylation of the tau protein. Inhibition of calpain, an enzyme showing increased activity in AD brains, led to promising results in animal studies. Calpain cleaves the CDK5 activator p35 leading to p25 formation and CDK5 overactivation.
Several acetylcholinesterase inhibitors such as tacrine, donepezil, rivastigmine, and galantamine have been approved for the treatment of mild to moderate AD by the FDA and other authorities. They act by reducing the deficits of the neurotransmitter acetylcholine associated with cognitive impairment in AD patients. The amantadine derivative memantine, an NMDA receptor antagonist, which was already used for the treatment of moderate to severe AD in Europe, has gained approval in the United States by the FDA as well.
A promising drug candidate, the β-secretase inhibitor verubecestat (MK-8931) developed for the management of mild to moderate AD, has moved to phase III. Moreover, the BACE inhibitor AZD3293 showed encouraging results in clinical studies. Antibodies as aducanumab and solanezumab, which have been designed to degrade plaques and lower the level of Aβ in the brain, have reached advanced stages of clinical testing for mild cases of AD.
Despite the many promising therapeutic approaches, AD still remains a major burden for the patients, their relatives, and the society.
| DOI | 名称 | |
|---|---|---|
| 10.1038/sj.mp.4001516 | Beta-sheet breaker peptide prevents Abeta-induced spatial memory impairments with partial reduction of amyloid deposits | 下载 |
| 10.1007/s00894-009-0594-y | Exploration of the mechanism for LPFFD inhibiting the formation of beta-sheet conformation of A beta(1-42) in water | 下载 |
| 10.1002/jmr.1113 | Effect of the beta-sheet-breaker peptide LPFFD on oriented network of amyloid β25-35 fibrils | 下载 |
| 10.1021/jp1116728 | Inhibition of aggregation of amyloid peptides by beta-sheet breaker peptides and their binding affinity | 下载 |
| 10.1038/nm0798-822 | Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy | 下载 |
| 10.1016/j.bbrc.2009.01.090 | Design and biological activity of beta-sheet breaker peptide conjugates | 下载 |